Journal Volume 1 - January 2006
Article 9
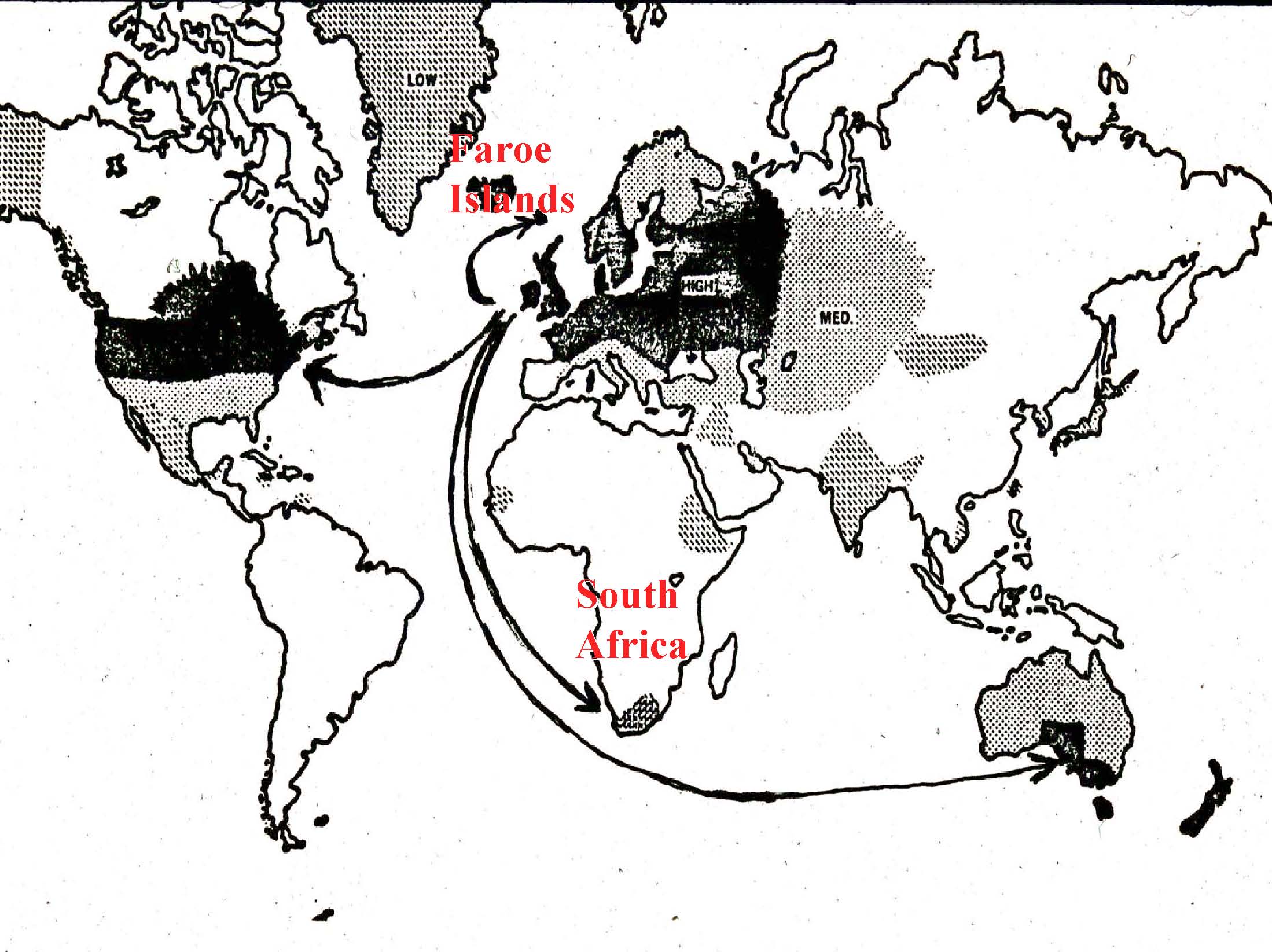
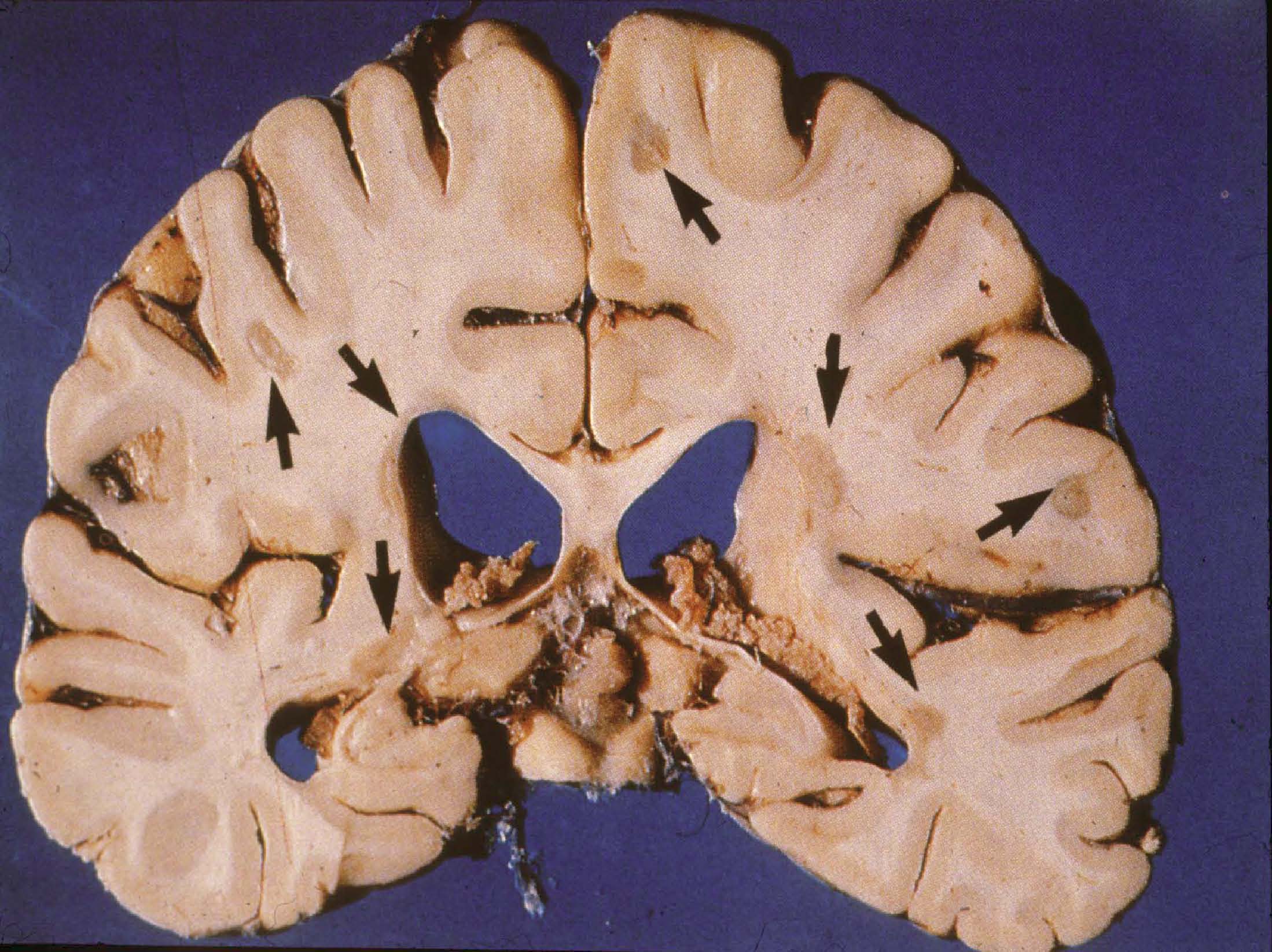
An Overview of Immunopathogenetic Mechanisms This paper will present you with some basic information about the composition and function of the immune system and how the immune system interacts with the nervous system. My clinical and research specialization is multiple sclerosis. I will use MS in this paper as the primary example to demonstrate some of the principles I will be presenting. It is useful to bear in mind that many of the things that happen in MS are very similar to what happens in TM and some of the other rare neuroimmunologic disorders. I am going to describe the epidemiology of MS – the how, what and why of this disease. I will focus on the immune cells and the primary players in MS; the T-Cells and the B-Cells and the macrophages. I will present you with information about some of the cytokines which are the hormones that are released into the spinal fluid which allow the cells to communicate with each other. I will describe how T-Cells and other cells become activated, and the mechanisms that account for why this whole autoimmune process happens in some people and not other people. I will talk about the stages of the MS lesion and different pathological variances. There is much we can learn from the fact that everyone presents in slightly differently ways; it is almost as if this is more than one disease process. As with MS, we also see lots of different variants of transverse myelitis and we are now starting to understand this variance at a pathological level. Finally, I will discuss the clinical correlations; the importance of bridging back and forth between what we see under the microscope (what we observe pathologically) and what we see with our patients (what we observe clinically). This important approach that connects the study of these diseases with the treatment of patients facilitates our learning more about these diseases and will ultimately help us devise better therapies for treating different subtypes of these diseases. With all autoimmune diseases and in fact with many medical illnesses in general, the manifestation or development of the disease is the result of an interplay of many different factors (figure 1). These factors include genetics; what we are born with and who we are that makes us individuals. It is an interesting situation that as scientists, we, unfortunately, spend a great deal of time studying clones of cells or strains of mice that are virtually identical in the laboratory. What we find in the laboratory does not always translate into what happens in the clinic, and that is because we are very different. We all have slightly different variations of our genes. The environmental factors that we all experience are also different. Some of us were raised in equatorial regions, and some in the far northern latitudes. Some of us are exposed to different viruses, different amounts of the virus and at different times in our lives. These variations in environmental experience may dictate, depending on a person’s genetic make-up, how one responds to the virus. And then the immune system has a role in the development of the disease. There is an interplay of all these factors that probably determines the different subtypes of these diseases. MS offers a good example of the influence and interplay of these factors, because we know that genetics (a genetic predisposition), the environment and immune factors are involved. We see, pathologically, and on the MRI, evidence for inflammation and damage to the myelin (demyelination). We also see this in TM. And then ultimately, if this process is vigorous enough or lasts long enough, we see damage to the underlying nerve wires, the axon fiber bundles that carry the signals from the brain down through the spinal cord and the sentry ones that go back up again. Turning to the pathogenesis of MS; pathogenesis is the medical concept for “how does it happen.” One of the prevailing theories in many of these autoimmune diseases is that there is some kind of infection. The infection is the environmental trigger that, through a process that we call molecular mimicry, or better thought of as “mistaken identity,” the immune system fights off the infection, then goes around looking for things that look like that infection. Then, if the infection is appropriately cleared, sometimes the immune system in people with autoimmune disease does not turn off properly and it will try to find something that looks a little bit like that infection. We know that there are things in the nervous system that actually have sequences of amino acids that are very similar to some of the common infections to which we are all exposed. One possibility is that we all get exposed to the mono virus, the Epstein-Barr virus, but some people do not turn off their response to it. Consequently, their T-Cells go around looking for things that look like that virus, and it turns out that there are fragments of myelin proteins that actually look a lot like that virus, and then those T-Cells may go in and attack the myelin. There are other possible mechanisms that might be involved that I will present later in this paper. Immune modulation is the most successful therapeutic approach to treatment of MS; this supports the role of the immune system in the pathogenesis (cause and development) of MS. We know that modulating the immune system, if we catch it early, is very effective in treating this disease. If damage to the axons occurs, however, then we need to think about other therapies, such as repairing or restoring myelin, or providing things to improve the function of the underlying nerve and the axon. There is continuing research on these forms of therapies and there is great potential for the future. This is the worldwide prevalence of MS (Figure 2). There are these geographic differences in the distribution of MS which can be interpreted in two ways. These differences could reflect changes in genetic pools. We know that MS is common in northern European populations and in northern parts of the United States, as well as in southern Australia and New Zealand. These locations represent common migration patterns; people from the UK have migrated to both places, as well as northern Europeans to the US. These differences could also reflect commonalities in environmental exposure. We know that in damp, cold places like Baltimore in the winter and the west coast, such as Seattle, and in the UK, that we see a lot of MS. It may be that exposure to viruses that are prevalent in these regions that are not prevalent in the equatorial regions, may increase the risk. When we look at the brains of patients with MS, we see areas of sclerotic plaques (Figure 3). In fact, the disease was described pathologically over a hundred years ago by French neurologist Charqeau as “le sclerose en plaque,” which basically means hardened grayish areas. We know today that the acute lesions are actually more inflamed; they can actually look reddish, as there are swollen blood vessels. So how does it happen? Well, the immune cells that are in your blood that are normally there to fight off infections actually somehow get into the nervous system. This image is a blood vessel (Figure 4). You can see red blood cells and white blood cells. The white blood cells are a part of the immune system that is designed to fight off infection. The white blood cells are somehow getting out of the blood vessel and they are going to areas around the blood vessel. In this case they are migrating into brain tissue; they are recognizing something there that they think they are supposed to be responding to or attacking. Again, perhaps they were thinking that they were finding the mono virus in the brain and what they are really seeing are myelin proteins that look like that virus. The immune cells in your brain start causing damage. They release cytokines, the communicating molecules that send activation signals to other cells. Some of these molecules are directly causing damage. The light grey area in this image is a myelin stain (Figure 5). Normally there would be an even pattern throughout the white matter. In this image there is a focal area that has been completely destroyed (white area). There are other less intense white areas that are partially, but not fully, repaired myelin. As I noted previously, the underlying nerve fibers become unhappy after this immune attack. In this image, we can see that some of these nerves have actually balled up (Figure 6). Imagine that if you cut a rope, or if a rope is frayed at the end, you will see the little fiber bundles end up in a little ball. This is what happens in these inflammatory lesions. We see this in MS, we see this in TM, and it has been described in other disease processes. Again, the axon loses some of its myelin, and then becomes balled up into these little forms that we call “spheroids.” The following is some very basic information about the immune system. Our immune system functions to protect us from infections; from bacteria, viruses, parasites, and fungi (Figure 7). The immune system protects us through two mechanisms. There is an innate immunity and an adaptive immunity. The innate immunity is what we are born with; we are born with some cells that can respond to infections immediately. They do not have to be trained or taught to do anything; they are scavenger cells and they can naturally kill some of the invading organisms. Our innate immunity is our first wall of defense. Then there is a second mechanism of immunity that becomes more activated in these autoimmune diseases. After your innate immune system either fails or the infection persists, then the adaptive immunity gets turned on. Another type of T-cells are called CD-4 or “helper” cells. They activate the CD-8 cells. They can also activate another part of our immune system, the humoral immune system or the B cells. We are interested in trying to specifically target these B lymphocytes from getting into the nervous system. One of the therapies that is showing a lot of promise, and may be the next approved drug for MS, is called “Natalizumab” or “Tysabri,” which targets a surface receptor on some of the T cells and prevents them from getting into the nervous system and causing this damage. The humoral immune system is boosted by TH2 (CD 4) cells or helper cells. These cells have been implicated in providing help to the B lymphocytes. The B lymphocytes are the cells that start making what are called immunoglobulins or antibodies, which can bind to different things and they can help the immune system then attack and kill a specific other cell. In the case of these inflammatory autoimmune conditions the antibodies bind to normal cell proteins like myelin or even perhaps the nerve fibers themselves and can cause damage through activation of some other proteins. There is a whole cascade of events that is extremely intricate and complicated. There are researchers who spend their whole careers trying to tease apart little surface receptors on these cells to try to ask questions like, “well what is it doing during this stage of MS, or what is it doing at this stage of transverse myelitis?” “Can we find something on the blood cells that will tell us that they are programmed to be going into the nervous system, so maybe we can prevent it from happening or prevent another attack from happening?” There is an entire discipline of study about the development and maturation of the cells of the immune system. Cells of the immune system begin life in the bone marrow as stem cells. We have stem cells in the bone marrow that are specifically designed to make new blood cells. They make red blood cells, and they make white blood cells. There are precursors to all of the different subtypes that make up the immune system and that we have been talking about. T-cells come out as a precursor from the bone marrow and go to the thymus gland which is located in the neck. T-cells are differentiated in the thymus; T-cells are “educated” in the thymus gland. The B-cells are differentiated or mature in the bone marrow. The B-cells then come out of the bone marrow and go into the lymph nodes and undergo a further maturational process. What the T-cells and B-cells share in common is that both end up in your lymph nodes, whether it be in your neck or under your arms; you have lymph nodes all over your body that are constantly responding to these different foreign invaders. This process is depicted in the cartoon (Figure 8). The T-cells go into bone marrow prep school, they come out and go to thymus university, and they learn what they are supposed to respond to. The thymus gland is a major focus of study in this process, because it is possible that this may be where the problems originate. A selection process occurs in the thymus. The T-cells are exposed to different antigens, cell proteins and foreign antigens. Basically, you want a repertoire of T-cells that respond to anything that might be seen. So we have thousands and thousands of different T-cells with little recognition modules called receptors that allow us to respond to all different sorts of infections. Some of those T-cells actually also respond to cell proteins, but the thymus is pretty clever and limits the number of those cells. But it turns out that we all have a small number of what we call “autoreactive” T-cells coming out of our thymuses. So, why is that? Well, it may have been Mother Nature’s clever way of allowing us a means to get into parts of the body where those T-cells normally shouldn’t go, but under extreme circumstances it might be beneficial. Normally we don’t get brain infections and we don’t want a lot of cells going into the brain, but it might be useful to have a few T-cells that actually do recognize some of the things that are in the brain; the myelin and the axons. Unfortunately, in the cases of infection, some of those T-cells may become expanded abnormally and start causing disease processes. But it is important that we understand why we have these cells and how we can keep them regulated or “tolerant” so that they are not attacking. They are just there as surveillance cells. They go in, they look around to see if there is any problem, and then if there is no infection or activation or what some people call a danger signal, they will actually leave the brain and go back out to the immune system. In the peripheral immune system where this little T-cell is going, where the action happens in the lymph nodes, we know that these foreign proteins get brought to the lymph nodes and the lymph nodes trap them. There are scavenger cells called macrophages that pick up foreign proteins and bring them to the lymph nodes and present them to the T-cells. If one T-cell comes along and says, “Ha, that is the one that I am supposed to be responding to and that is my mission in life,” then it goes off and attacks that protein and in some cases, as I said, some of those T-cells are actually programmed to go into the brain. Another important area of study concerns how T-cells become activated (Figure 9). There are signals that occur and the little protein (depicted as the irregular circular mass where the antigen-presenting cells are located) has to get presented by little surface molecules on the macrophages. The T-cell recognizes it in the context of its little T-cell receptor. There are other communicating molecules, and there is an initial activation process. In autoimmune diseases we think that a marker of the cells might be chronic activation, or a chronic stimulation and differentiation of a different cell type that has a different set of surface receptors. We are now getting to the point in our understanding of the process where we can take blood cells out of the patient with MS or some of these other diseases and see differences in their surface receptors that allow us to say which of the thousands and thousands of white blood cells are the ones that are being chronically activated. Although we may not yet know which are the actual antigens that we are responding to, we have narrowed it down from hundreds of thousands of cells to maybe a thousand or so cells that look like they are abnormal in some of these diseases. Of course, that is really where we need to be going, because we need to have targets. Imagine, we have all these T-cells and we want to find the ones with the red hats; and it turns out that there are probably about one in ten thousand. With some of the sophisticated equipment that we have in our laboratories, we can pull out that one cell from among the ten thousand, and then we can devise strategies to specifically target it. And that is really where the whole field of immunology is headed. There may be another approach that can be employed in treating these autoimmune diseases. If we are not able to find the specific T-cell that is initiating this process, it might be possible to block some of the communicating cytokines that are released by these cells. I previously described the TH1 and TH2 type cells. There are different cytokines that are released by these different cell types and they provide different functions. In MS, we think that the TH1 cytokines, interferon gamma and tumor-necrosis factor are the bad guys. In transverse myelitis it may be different. Some studies suggest that the TH1 cells may be the culprits in MS. But in these very hyper-acute or very immediate attacks on the nervous system (as in transverse myelitis) you can get TH2 responses. This is very important, because some of the strategies in MS to block these cytokines may actually not work in these more acute situations. Some of the TH2 median diseases also may have an antibody component, and blocking the cytokines might not be enough. We may need to do a blood-washing procedure called plasma exchange that pulls out some of the antibody proteins that are circulating in the blood of people who have this kind of presentation. This is a very busy image that demonstrates the very complicated process that I have been describing (Figure 10). What is depicted in this diagram are cells that are flowing in the blood stream or in the blood vessel. The cells of the blood vessel wall compose what we call the blood-brain barrier. Cells that get to the other side of the barrier, in the brain or spinal cord, can cause a whole series of activation events; activating other cells that can destroy the myelin around the nerve or break up the nerve itself. As immunologists, we are constantly looking for these different targets; we are looking for targets directly on the T-cell. We are looking for some of the receptors that allow it to stick to the blood vessel wall and we are looking for ways to block the cytokines. It is really important that we find out which are the bad guys in a specific disease process and how much it might differ between one person and another. And again it is this common theme of variability in the response; finding the answer to the nature of this variability that is going to be absolutely critical as we try to tailor our therapies towards the specific sub-types of the disease. There are a number of potential mechanisms of autoimmunity. I described one mechanism, molecular mimicry, in which there is mistaken identity between viruses and normal cell proteins. How else can infections and the environment activate our immune system? There are some infections that release substances that activate immune cells either directly or in a non-specific or indirect manner. A good example of this is either food poisoning or toxic shock syndrome. In those cases we get exposed to bacteria or sometimes viruses that release substances called super-antigens that can bind to the T-cell and just activate it. As I noted previously, we all have these low levels of auto-reactive T-cells that are supposed to be just there undergoing surveillance. Well, if you get a bad infection, for instance a bacterial infection like toxic shock, it releases these super-antigens, and it has been shown in some models now in isolated clinical cases, that those infections can activate your autoimmune cells to go in and cause some damage. One would think that once the infection was cleared that the process would turn off, and that may well be what happens. In fact, that may be why we see some neurologic diseases that are just one time episodes where it does not relapse and remit the way MS does. We know, for example, that transverse myelitis for most people is a one time episode. If we could identify who has those super antigens early on, we might be able to limit the damage. So, the process does not just occur like a stroke within an hour; it probably occurs over a few days to weeks. There is some very exciting research that is attempting to identify super-antigens early in the intensive care unit setting and trying to devise drugs that might block this super-antigen activation and limit the inflammatory damage right when the patient is coming in with their presentation. Inflammation from infection causes release of self proteins. Normally, most of your brain proteins are isolated in your brain. We do not have a lot of fragments of our brain in other places, which is a good thing. But when you get an infection, sometimes those proteins get released, they drain into the lymph nodes in the back of your neck, and then the immune system has more exposure to them. Sometimes proteins in the myelin sheath that are not ordinarily exposed because they are all wrapped up tightly may become exposed. When the myelin sheath gets disrupted from a one time event, such as from a traumatic event, this causes a release of self proteins that normally are on the inside of the myelin sheath where your immune system is not able to see them. Suddenly the immune system sees them and says, “Uh-oh, danger, there is something wrong.” It thinks that it might be an infection and it starts attacking it. So inflammation and damage of tissues can cause release of these self proteins. A final mechanism that has been shown to occur in some diseases is the direct infection of the lymphocytes, either the T lymphocyte or the B lymphocyte. We know in one particular disease that causes a spinal cord disorder mediated by a retro virus called HTLV-1, that the virus can infect the T-cells and causes those T-cells to just start dividing rapidly and making their own cytokines. The virus itself activates the cells. And it turns out that in other viral infections like the mono virus, EBV, this virus can get into your B cells and causes the B cells to do funny things, such as divide and release cytokines. Thus, direct infection may be something that happens as well. There are other factors that are involved in this process. Genetic susceptibility is absolutely critical. We need to be doing wide screening of people to try to understand what are the genes that predispose people to these aberrant responses. There may be an important role for hormones in this process. We need to understand why a lot of autoimmune diseases are more common in women; we need to develop a better understanding of the role of hormones. Why in MS does the disease becomes quiet during pregnancy and then exacerbates post partum. In lupus, however, the disease is aggravated during pregnancy and is actually better afterwards. For instance, it is possible that there is a shift of these TH1 TH2 cytokines driven by some of the female hormones, like Estriol. It turns out that there are programmed cells to go to specific sites of the body (Figure 11). Some people may be susceptible to autoimmunity. But one might ask, “Why is it that my Grandmother got rheumatoid arthritis and why do I have MS?” It may be that there is a gene or set of genes that predisposes to autoimmunity, but there are other things that then tell the cells to go to the joints in rheumatoid arthritis, or go to the gut in Crohn’s Disease, or go to the pancreas in diabetes, or go to the nervous system in MS and TM. So we are trying to understand the combination code for entry into different tissues; skin, the mucous, the gut or the brain, and although it turns out that they probably are not specific codes, that there are patterns that do define the sites for migration. So, just like a key, it is not going to be one notch, but it is going to be a very complicated pattern that tells the cell, you need to go to this specific site. The cells migrate across the nervous system, there are different signals (Figure 12). We are interested in chemokines, which is a kind of cytokine that basically sends out a signal and says, “Hey guys, come over here, this is where the action is.” Maybe blocking those will be an important therapy. As I said, there are different responses to this inflammation. In MS, we recognize four different types of pathologies that in many ways determine what happens to the patients (Figure 13). One of the great distinctions in the pathologies between types one and two, and types three and four is that the myelin-making cells that we call oligodendrocytes die in type three and four, and there is very little remyelination. We have recognized this clinically. Some people have attacks and recover well and other people do not. We have been trying to understand this observation on a pathological basis, studying tissues under a microscope. There is a need to go back to the clinic and try to figure out the subtypes of disease; we need to identify measures of disease activity, not in the tissues, but perhaps using blood samples, spinal fluid samples or MRI (Figure 14). I would like to briefly mention the stages of MS, which are also important (Figure 15). Some of the inflammation happens early. We see relapses; we see an active disease on the MRI. The MRI tells us that in the late stages of MS, there is more degeneration of the nerves and the nerve wires. Disability gets worse during this stage and there is a “different” sequence of events on the MRI’s. The conventional MRI is a window into the brain and spinal cord and does give us very interesting and important information about how we might focus treatments (Figure 16). When there is a lot of inflammation, we might want to focus our treatments on suppressing the inflammation. In the later stages when the inflammation is less, and there is more tissue damage, we might want to focus on restoring nerve function. These are some of the beautiful pictures that we can now get; we can see areas of demyelination in the deep parts of the brain (Figure 17). We can see areas where we have injected a dye called gadolinium. The lesions enhance just like the ones in the spinal cord do that you may have seen in transverse myelitis. The demyelinating and inflammatory lesions in the brain are enhanced, so we know that these are very active lesions and we should be targeting the inflammation here. But remember in this case, those cells are already in the brain, so we need something that will actually get into the brain to help stop them. And we know that in this stage of the disease with these darkened areas of scar tissue that the nerve fibers have dropped out. When we start to see these, the patient might need neuroprotective therapy to keep their nerve cells happy, so they don’t start dying out, like in the very dark area. In the whole field of autoimmunity we need to be thinking about the stages of disease, the types of disease, and combinations of therapies (Figure 18). These diseases are not progressing in distinct stages; there is a series of events that happen slowly. We know that although inflammation happens early in MS and the axonal damage ensues afterwards, you can actually see some axonal damage right at the outset. We need to target the immune system and we need to target protecting the nervous system simultaneously. In summary, MS and many of these other neuroimmunologic diseases are inflammatory and what we call degenerative; so the nerves become unhappy and die. The future is in diagnostic testing to better classify subtypes of disease using blood, spinal fluid and MRI. I am very hopeful and optimistic that we are headed towards a time where we can tailor our therapies to the subtypes of disease and the stage of disease; and I think that we are already starting to do that right now. |
{kind=link}
{kind=link}
{kind=link}